带隙计算公式 npj: 准确计算能带带隙—WKM方法
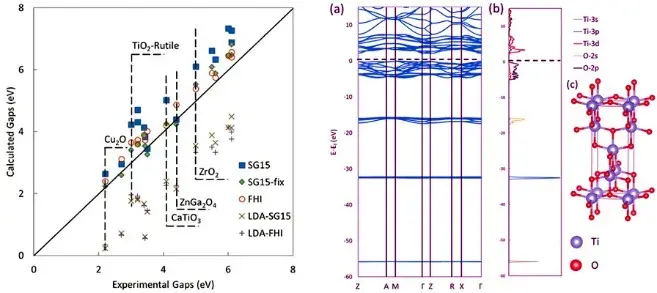
使用普通平面波Kohn-Sham方程计算的密度泛函理论(DFT)时,通过理论计算得到的带隙往往会小于实际实验得到的带隙。一般而言,DFT+U可以通过调整U值、杂化密度泛函可以通过调整杂化比例,得到与实验相符的带隙,但是这些计算方法需要有经验依据来设定参数。DFT+U在线性响应的计算方法可以得到符合Koopmans 定理的U值,并能得到合适的带隙,但是这样的方法不能用于闭壳系统。GW计算方法会消耗大量计算资源,根据其不同的收敛特征可分为G0W0、GW0、GW等计算方法,而完全收敛的GW计算则往往得到比实验值更大的带隙。WKM(Wannier-Koopmans Method)是一种基于Koopmans定理、wannier函数以及常见的交换关联泛函(如LDA、GGA等方法)的计算方法。它在半导体、离子晶体、有机共价晶体和二维材料等计算中都可得到与实验一致的带隙结果。因此,该计算方法是否有可能在不大量增加计算时间的情况下得到准确的电子结构?
来自美国的劳伦斯伯克利国家实验室的汪林望博士与北京大学深圳研究生院的潘锋教授合作,使用WKM方法针对过渡金属氧化物作了研究。他们在计算中发现,通过去除交换关联泛函中的d轨道wannier函数与内层电子的相互作用,即可在d0和d10闭壳系统中得到与实验一致的带隙大小。而对于开壳系统的过渡金属氧化物,由于d轨道中占据的wannier函数与非占据的wannier函数间相互作用,WKM目前还无法计算得到与实验一致的带隙结果,但通过分析,本工作为后续改进WKM方法提供了新思路。
该文近期发表于npj Computational Materials 6: 33 (2020),英文标题与摘要如下,点击
https://www.nature.com/articles/s41524-020-0302-0可以自由获取论文PDF。
Wannier Koopmans method calculations for transition metal oxide band gaps
Mouyi Weng, Feng Pan, Lin-Wang Wang
The widely used density functional theory (DFT) has a major drawback of underestimating the band gaps of materials. Wannier–Koopmans method (WKM) was recently developed for band gap calculations with accuracy on a par with more complicated methods. WKM has been tested for main group covalent semiconductors, alkali halides, 2D materials, and organic crystals. Here we apply the WKM to another interesting type of material system: the transition metal (TM) oxides. TM oxides can be classified as either with d0 or d10 closed shell occupancy or partially occupied open shell configuration, and the latter is known to be strongly correlated Mott insulators. We found that, while WKM provides adequate band gaps for the d0 and d10 TM oxides, it fails to provide correct band gaps for the group with partially occupied d states. This issue is also found in other mean-field approaches like the GW calculations. We believe that the problem comes from a strong interaction between the occupied and unoccupied d-state Wannier functions in a partially occupied d-state system. We also found that, for pseudopotential calculations including deep core levels, it is necessary to remove the electron densities of these deep core levels in the Hartree and exchange–correlation energy functional when calculating the WKM correction parameters for the d-state Wannier functions.